
Cleaning up SRM data in Skyline, part II
Before I export my data from Skyline for data analysis, I have the following final things to do:
- Adjust peak boundaries as necessary
- Remove transitions that do not align with the predicted RT. Remove any peaks that do not have at least 2 transitions.
Some helpful keyboard shortcuts:
- Scroll between replicates: Ctrl+Up or Ctrl+Down
- Auto-zoom to best peak: F11
- Un-autozoom out from best beak: Shift+F11
Some transition peaks look poor, but they are present. Here is an example of replicate data for the Superoxide Dismutase protein, showing the overall view and the zoomed view:


This is in comparison to the following replicate, where there is no peak present betwen RT 14-15:


Not sure what to do in the situation where a peak is split into 2 peaks (as below); Skyline opted for the boundaries to encompass both peaks. I will do the same, as the total RT for both peaks appears to be similar to that of other reps:

Notes:
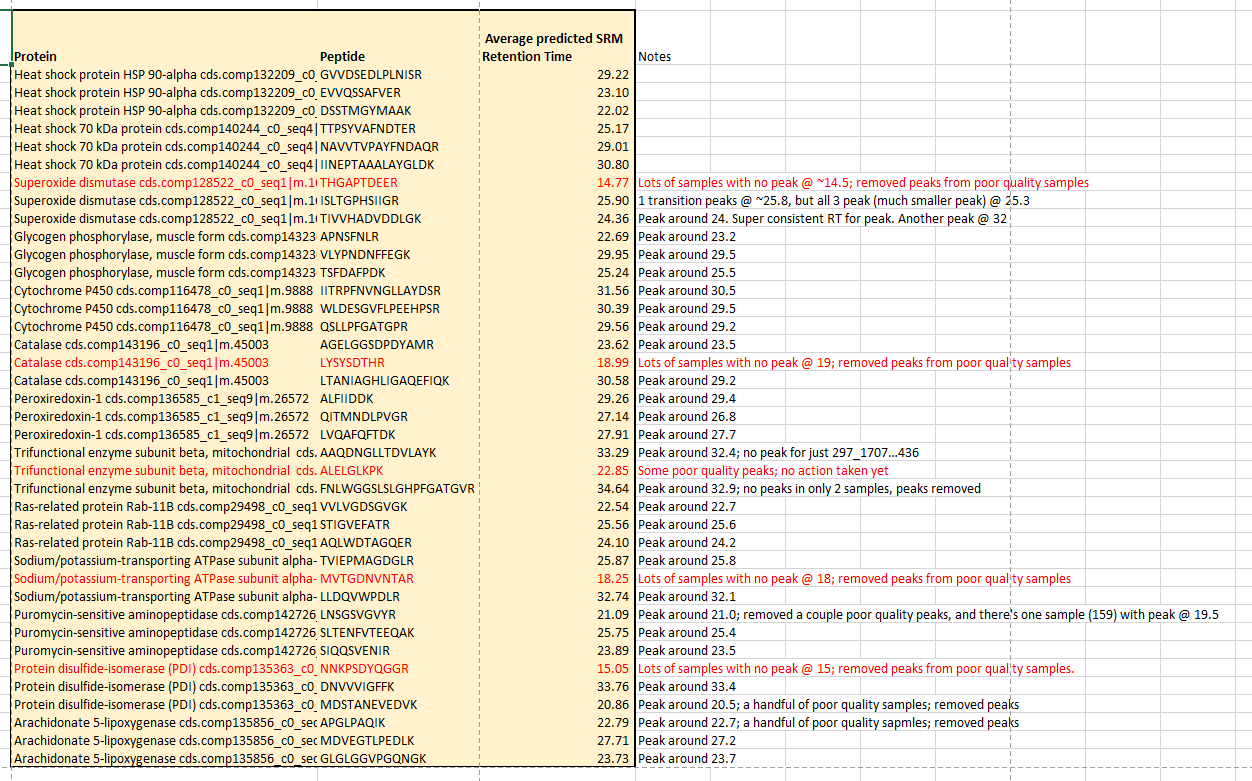
- Poor quality reps: 178, 254, 208, 212, 213, 297_170728020436,
- A peptide with RT ~18 must be co-eluting, as it pops up in a few res/peptides. Perhaps it is the [PEPTIDE W/ 18], that gets stuck in the column and pops up. For example, the following is a zoomed-out view of Ras-related protein peptide, which should have it’s peak around 22.7. This is rep #254, but it pops up in lots of reps:

 A couple peptides elute @ ~18min, and could be the culprit: Sodium/potassium-transporting ATPase subunit alpha-4, MVTGDNVNTAR; Catalase, LYSYSDTHR;
A couple peptides elute @ ~18min, and could be the culprit: Sodium/potassium-transporting ATPase subunit alpha-4, MVTGDNVNTAR; Catalase, LYSYSDTHR;
Total actions performed on SRM data in Skyline:
- Removed peaks from replicates where no peak was present @ designated retention time (as per DIA/SRM regression). Replications w/ no peaks for peptides will be represented as #N/A when data is exported.
- ID’d peptides with very poor data across multiple replicates; I may not use these peptides in my analysis; TBD. See peptides in red
- Adjusted retention time boundaries for all reps all peptides. I erred on NOT adjusting boundaries if they looked OK to maintain consistency.
- ID’d and deleted 2 transitions that do not align with other transitions at designated RT. Transitions are:
- Superoxide dismutase, TIVVHADVDDLGK, y4
- Ras-related protein Rab-11B VVLVGDSGVGK, y4
- Documented peptides and transitions w/ poor quality over several samples and saved in my Geoduck-DNR Analysis Repo
{kind=link}
Exported data from Skyline:
Export -> Report, then I edited the Transition Results report with the following metrics: Protein Name, Transitions, Peptide Sequence, Fragment Ion, Peptide Retention Time, Area; I selected “Pivot Replicate Name”. Here’s a preview of the report:

I then exported the same report, NOT pivoted by replicate name.
Then, I modified the report to remove retention time, and exported in both the pivoted and not-pivoted formats.
All files were uploaded to my Geoduck-DNR/Data repo:
SRM Transition Results, pivoted
SRM Transitoin Results, not pivoted
SRM Transition Results, no RT, pivoted
SRM Transition Results, no RT, not pivoted
Next steps:
- Determine if any .raw files should be discarded, based on poor-quality data and those that I re-ran & re-made
- Look @ all blank runs to see if there are any weird signals that linger
- Check out biological blank (sample prepped as per the protein extraction process, but had no tissues) to see if there is any contamination. Should also check out Yaamini’s blanks.
- Normalize based on PRTC peptides
- Review Dilution Curve results
- Generate NMDS plot
- ….