
Mass spec hiccup?
SPOILER ALERT The thought is that some of the peptides (in both PRTC and my samples) degrade quickly, and this is what’s causing the loss of signal. I loaded my samples in batches of 25, so samples sat out up to ~2.5 days prior to being injected. I’ll need to consider “time out of freezer” as a factor when analyzing my results. This is obvious in the dilution curve data, where PRTC total area shows an obvious decrease over time.
Things appeared to be going very smoothly.

Then, @ 3pm on Friday 7/21 I noticed my QC data beginning to look a bit off starting @ QC38:

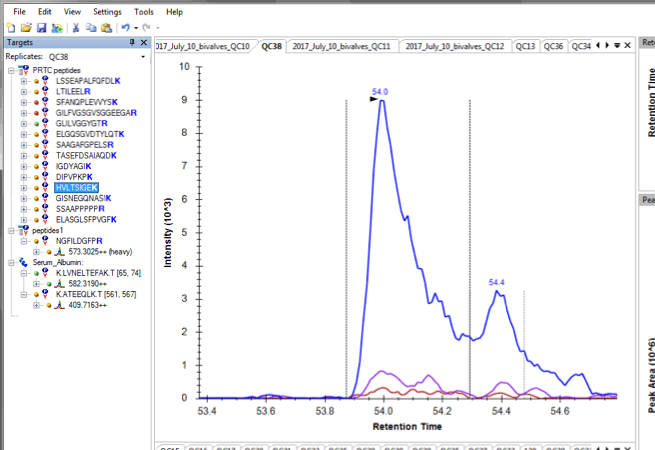


Consulted with Yaamini. She suggested checking out the PRTC data in my sample files. I loaded them into the QC Skyline file (PRTCplusBSA), and saw some oddities, particularly a handful of samples that had some completely undetected PRTC peptides. These samples are:


Consulted with Brook. These are her thoughts:
- If the other QC peptides look ok- it may be a factor that your column is retaining some interfering peptides that co-elute or are more dominant in your sample than the QC- and they, as a result, cloud the PRTC signal”
- You could do a few things-
- Run 2 blanks to clean things up again.
- Then do 3 qc’s and see if it all looks ok.
- Or you could re-run one of Yamini’s samples and see if it looks the same
- If Yaminis sample looks different on round 2, then something is up with the column
- if it is fine, something is up with your samples.”
I decided to run 2 blanks, then 3 QC’s like she suggests.
Also, I asked Brook this:
- Just a thought: Yaamini’s samples, the blank, and the QC all were prepared using 3% Acetonitrile + 0.05% Formic Acid by accident, while I prepared my samples with the 0.1% FA called for in the protocol. Is it possible that the change between FA concentration is throwing things for a loop? This just crossed my mind, since I’m about to add ACN+FA to the QC vial and am unsure which one (.1% or .05% FA) to add…
… and her response: - you want 0.1% formic acid- but that shouldn’t matter too much- not enough to make signal GONE
I made a new QC vial by adding 30ul to the 10ul QC aliquot I got from Emma’s lab, vortexted, spun it down a second, then transferred it to an autosampler vial and placed it in the tray @ 1:A,4
A couple more samples ran before I was able to queue up the blanks. Oddly enough, they look fine. The latest QC does not, though:


I’m going to run the 2 blanks, then 2 QC’s. Hopefully they look good. If not, I’ll have to run down to UWPR and put one of Yaamini’s samples in, run that, and see if that looks good or bad. If bad, then something is wrong with the column and Brook will meet me on Saturday morning to change it. If good, then something is wrong with some of my samples. Although, the samples that don’t look good are no different than the ones that look good that are adjacent- same PRTC, same preparation date & time, same time sitting out of the freezer, and a mix of Round 1 & Round 2 extractions, etc. So, I can think of no major difference between these samples that would cause such signal issues.


Here’s “bivalve_199”, which is the first blank in the flush

OK, after running 2 blanks here’s the first QC, “QC41”

The blanks and QC’s looked good, so I re-ran a couple of my poor-quality samples. They did not look good. So, the thought is something is happening in those samples, like degrading peptides, or co-eluting peptides. I decided to move forward with my other samples, and keep track of those that do not look good. I can then either re-run them, or remake and re-run depending on time.