
Post Skyline Data Processing
Please see my Geoduck-DNR Repo, June Analyses folder for most up-to-date version of this process.
In this workflow I process Skyline data to assess proteins expressed in Geoduck during outplant in 5 sites throughout Puget Sound in 2 treatments: eelgrass, and bare (no eelgrass).
Pertinent files for ths process: R code, Biostats.R
Step 1: Export report from Skyline
screen shots TBD
Step 2: Add site/treatment to columns, remove #N/A, and normalize based on Total Ion Current
The raw file from Skyline, opened in Excel, looks like this:
I used Excel to make these small modifications. Yes, I could have dones this in R, but I didn’t (next time I will). I referenced my Mass Spec Sequence File to match file names to sample numbers, and then my Geoduck Organization file to match sample names to site and treatment. To edit all #N/A I used the Find & Replace tool.
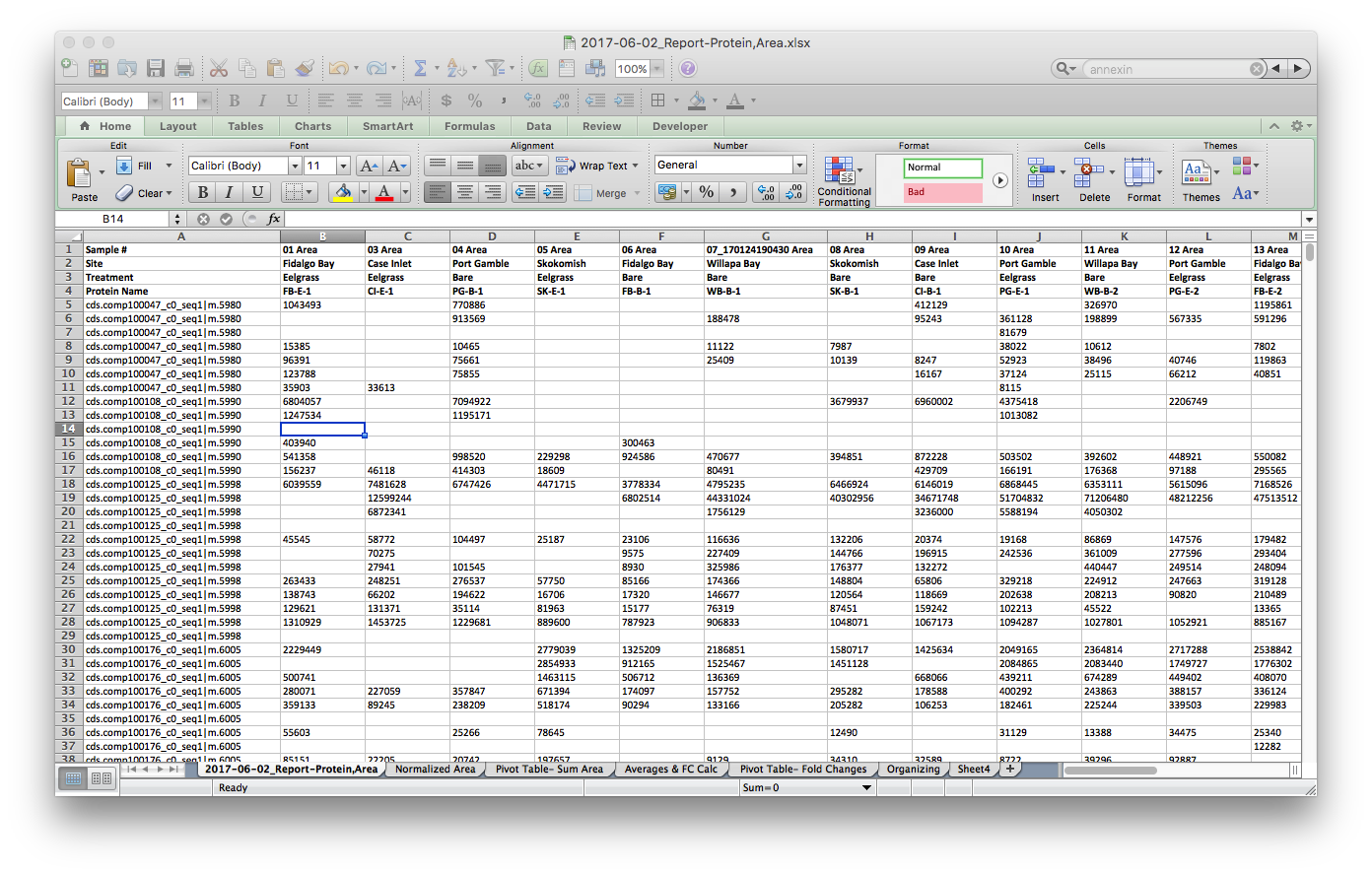
Finally, I opened a new tab and normalized the data by the Total Ion Current (TIC) for each sample. TIC data was given to me from Emma.
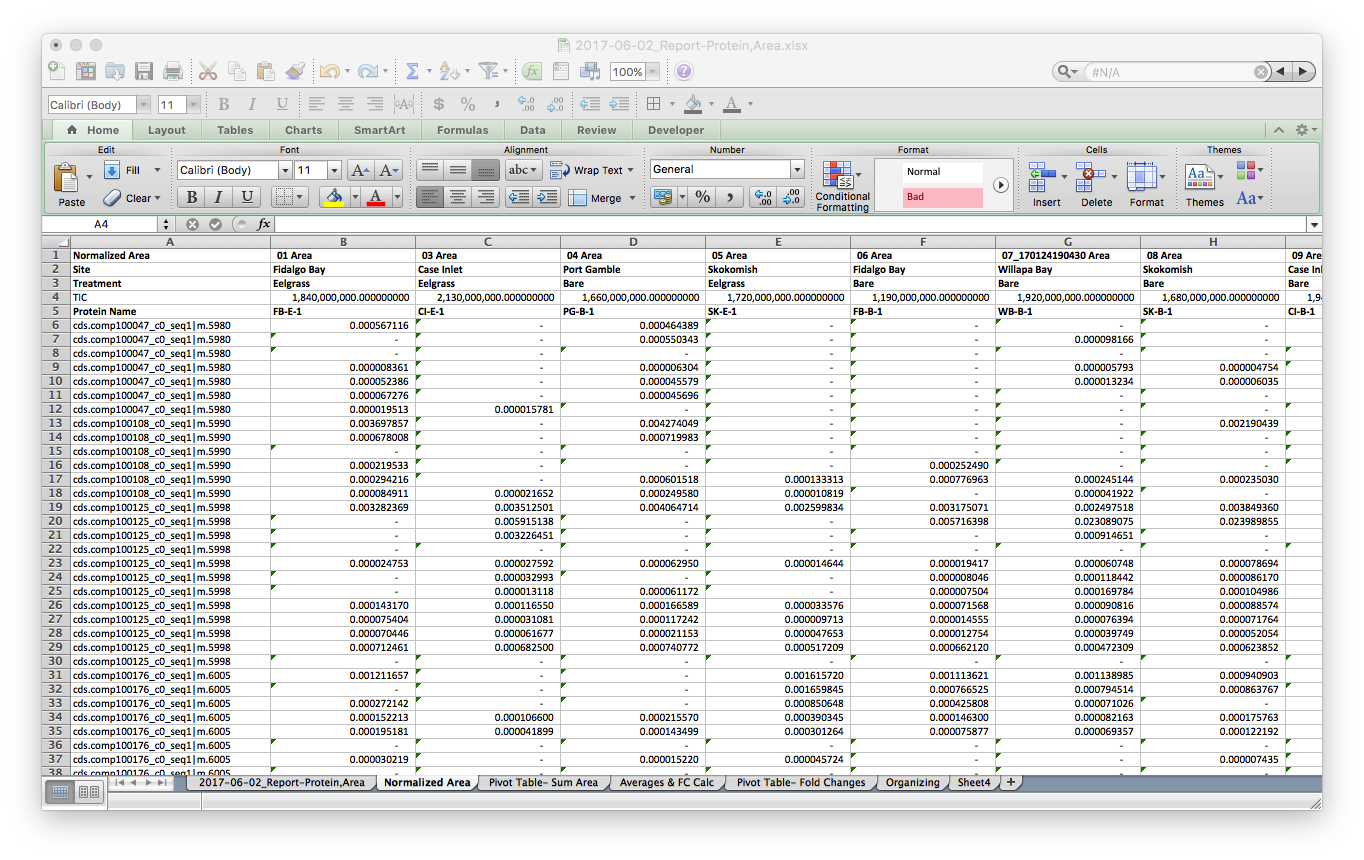
I saved this tab as a new file, 2017-06-02_SKYLINE-Total-Protein-Area-NORM.csv2017-06-02_SKYLINE-Total-Protein-Area-NORM.csv, cleaned up the header with simplified sample codes: e.g. FB-E-1 & CI-B-2 for Fidalgo Bay Eelgrass technical rep 1, Case Inlet Bare technical replicate 2, respectively. Also notice that all the n/a spaces are showing up as 0. I’ll take care of this in R
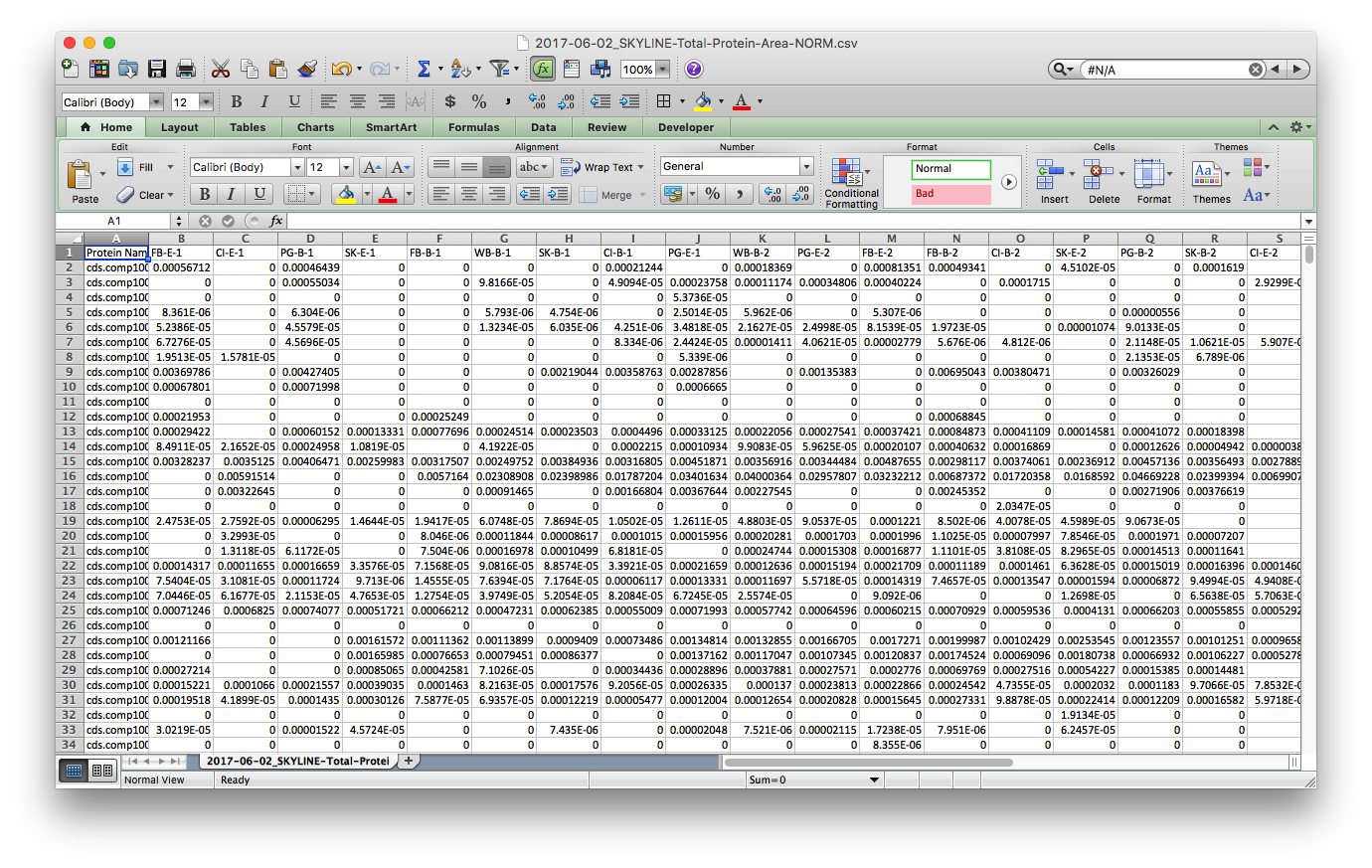
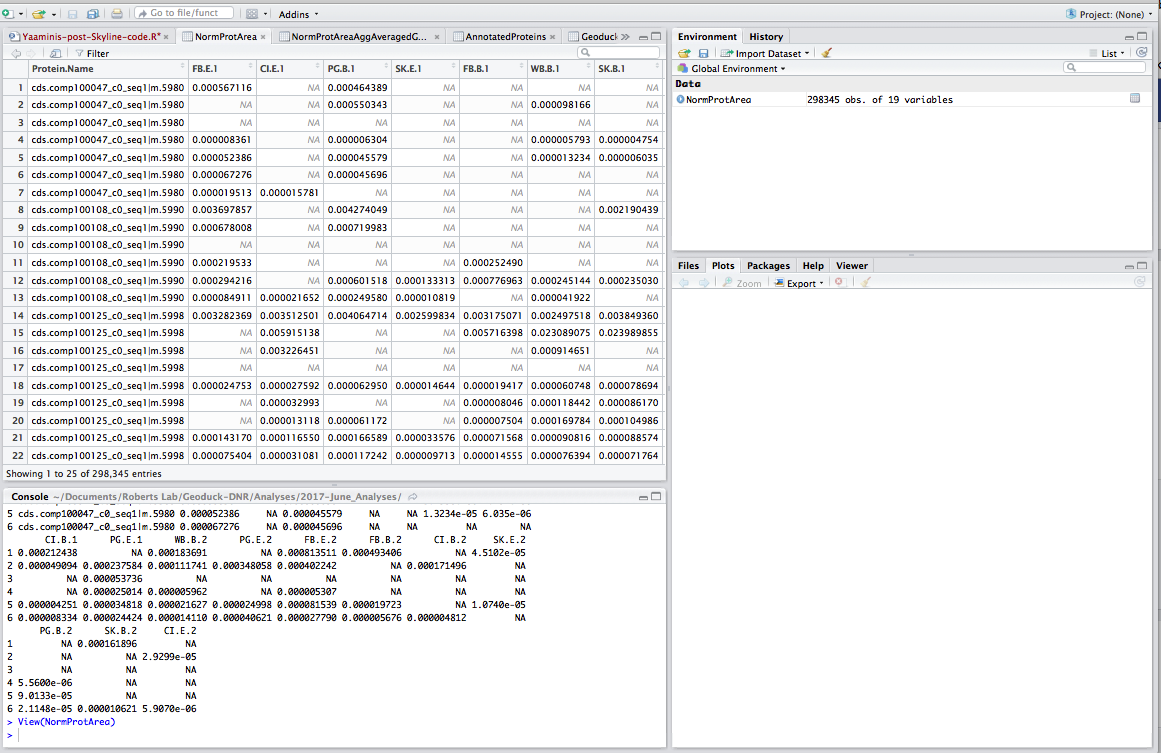
Step 3: Read data into R and play with it. I borrowed a lot of this script from Yaamini, with some modifications - thanks Yaamini!
### Import Dataset ###
### Note: this dataset has already been massaged in Excel: sum area by protein, normalized by TIC, all n/a's removed (replaced with zeros), and columns renamed
setwd("~/Documents/Roberts Lab/Geoduck-DNR/Analyses/2017-June_Analyses")
NormProtArea <- read.csv("2017-06-02_SKYLINE-Total-Protein-Area-NORM.csv", header=TRUE, na.strings = "0") # import local file
head(NormProtArea)
Step 4: Aggregate transition area by protein. Data currently represents the area under the curve for each transition. To find the total area per protein I use the aggregate() function to sum all transition areas by protein.
### SUM TRANSITION AREA BY PROTEIN ###
# The "Area" values (which have already been normalized by TIC) are peak area for each transition. Sum them to determine total area for each protein
NormProtAreaAgg <- aggregate(cbind(FB.E.1, CI.E.1, PG.B.1, SK.E.1, FB.B.1, WB.B.1, SK.B.1, CI.B.1, PG.E.1, WB.B.2, PG.E.2, FB.E.2, FB.B.2, CI.B.2, SK.E.2, PG.B.2, SK.B.2, CI.E.2) ~ Protein.Name, FUN = sum, data = NormProtArea, na.rm = TRUE, na.action = NULL)
head(NormProtAreaAgg) #confirming that proteins are now summed
Step 5: Average technical replicates. Each sample ran through the MS/MS twice; here I calculate the average for each treatment/site combo:
#### AVERAGE REPLICATES ####
# FYI No eelgrass sample for Willapa Bay
bareCaseInlet <- ave(NormProtAreaAgg$CI.B.1, NormProtAreaAgg$CI.B.2) # create vector for each site/treatment that averages the replicates (of which there are 2)
bareFidalgoBay <- ave(NormProtAreaAgg$FB.B.1, NormProtAreaAgg$FB.B.2)
bareWillapaBay <- ave(NormProtAreaAgg$WB.B.1, NormProtAreaAgg$WB.B.2)
bareSkokomishRiver <- ave(NormProtAreaAgg$SK.B.1, NormProtAreaAgg$SK.B.2)
barePortGamble <- ave(NormProtAreaAgg$PG.B.1, NormProtAreaAgg$PG.B.2)
eelgrassCaseInlet <- ave(NormProtAreaAgg$CI.E.1, NormProtAreaAgg$CI.E.2)
eelgrassFidalgoBay <- ave(NormProtAreaAgg$FB.E.1, NormProtAreaAgg$FB.E.2)
eelgrassSkokomishRiver <- ave(NormProtAreaAgg$SK.E.1, NormProtAreaAgg$SK.E.2)
eelgrassPortGamble <- ave(NormProtAreaAgg$PG.E.1, NormProtAreaAgg$PG.E.2)
NormProtAreaAggAveraged <- data.frame(NormProtAreaAgg$Protein.Name, bareCaseInlet, bareFidalgoBay, barePortGamble, bareSkokomishRiver, bareWillapaBay, eelgrassCaseInlet, eelgrassFidalgoBay, eelgrassPortGamble, eelgrassSkokomishRiver) # combine site/treatment vectors into new dataframe
head(NormProtAreaAggAveraged)
Step 6: Prep file to merge with annotations. The Protein ID in my Skyline files are formatted differently from the GeoID in my annotation file. Here I edit out the extra string (in bold), cds.comp100047_c0_seq1|m.5980
### EDITING GEOID NAME TO MATCH ANNOTATED FILE ###
NormProtAreaAggAveragedGeoID <- NormProtAreaAggAveraged # copy database, create new one to be used to join to annotated file
GeoID.a <- (gsub("cds.", "", NormProtAreaAggAveragedGeoID$NormProtAreaAgg.Protein.Name, fixed=TRUE)) # remove cds. from protein ID
GeoID.b <- (gsub("\\|m.*", "", GeoID.a)) # remove |m.#### from protein ID
noquote(GeoID.b) # remove quotes from resulting protein ID
NormProtAreaAggAveragedGeoID[1] <- GeoID.b # replace the newly created vector of protein ID's in the full database
head(NormProtAreaAggAveragedGeoID) # confirm changes
nrow(NormProtAreaAggAveragedGeoID) # confirm # rows still same
write.table(NormProtAreaAggAveragedGeoID, "2017-06-30_All-Proteins.tab", quote=F, row.names = F, sep="\t") # Save to file
Step 7: Upload annotations. This file is the annotated proteins found via geoduck gonad transcriptome.
### UPLOAD ANNOTATED GEODUCK PROTEOME FROM URL ###
GeoduckAnnotations <- read.table(
"https://raw.githubusercontent.com/sr320/paper-pano-go/master/data-results/Geo-v3-join-uniprot-all0916-condensed.tab",
sep="\t", header=TRUE, fill=TRUE, stringsAsFactors = FALSE, quote="") #fill=TRUE for empty spaces
nrow(GeoduckAnnotations) # check that the # rows matches the source (I know this from uploading to Galaxy; not sure how else to do it)
ncol(GeoduckAnnotations) # check that the # columns matches the source
head(GeoduckAnnotations) # inspect; although it's easier to "view" in RStudio
Step 8: Merge the 8076 total proteins found in my samples with the annotations. After merge, there are 5,690 annotated proteins in my data. Annotated file is 2017-06-30_All-Annotated-Proteins.tab
# MERGE ANNOTATIONS WITH ALL MY PROTEINS ###
AnnotatedProteins <- merge(x = NormProtAreaAggAveragedGeoID, y = GeoduckAnnotations, by.x="NormProtAreaAgg.Protein.Name", by.y="GeoID")
head(AnnotatedProteins) #confirm merge
nrow(AnnotatedProteins) #count number of proteins that were matched with the list of annotations
write.table(AnnotatedProteins, "2017-06-30_All-Annotated-Proteins.tab", quote=F, row.names = F, sep="\t") #write out .tab file
Step 9: Make NMDS plot to assess overall site relatedness. Again, thanks to Yaamini for providing me the code! Note: I saved the biostats.R code as an R script in my repo/working directory
#### CREATE NMDS PLOT ####
#Load the source file for the biostats package
source("biostats.R") #Either load the source R script or copy paste. Must run this code before NMDS.
library(vegan)
#Make sure first column of protein names is recognized as row names instead of values - this takes the first column containin protein names, and assigns it to the row "header" names
area.protID <- NormProtAreaAggAveraged[-1]
rownames(area.protID) <- NormProtAreaAggAveraged[,1]
head(area.protID)
write.csv(area.protID, file="2017-07-04_NormProtAreaAgg&Ave.csv") #write this file out for safe keeping
#Transpose the file so that rows and columns are switched
area.protID.t <- t(area.protID[,1:9])
nrow(NormProtAreaAggAveraged) # Compare row and column lengths before and after transposing
ncol(NormProtAreaAggAveraged)
nrow(area.protID.t)
ncol(area.protID.t)
#Make MDS dissimilarity matrix
proc.nmds <- metaMDS(area.protID.t, distance = 'bray', k = 2, trymax = 100, autotransform = FALSE)
#Make figure
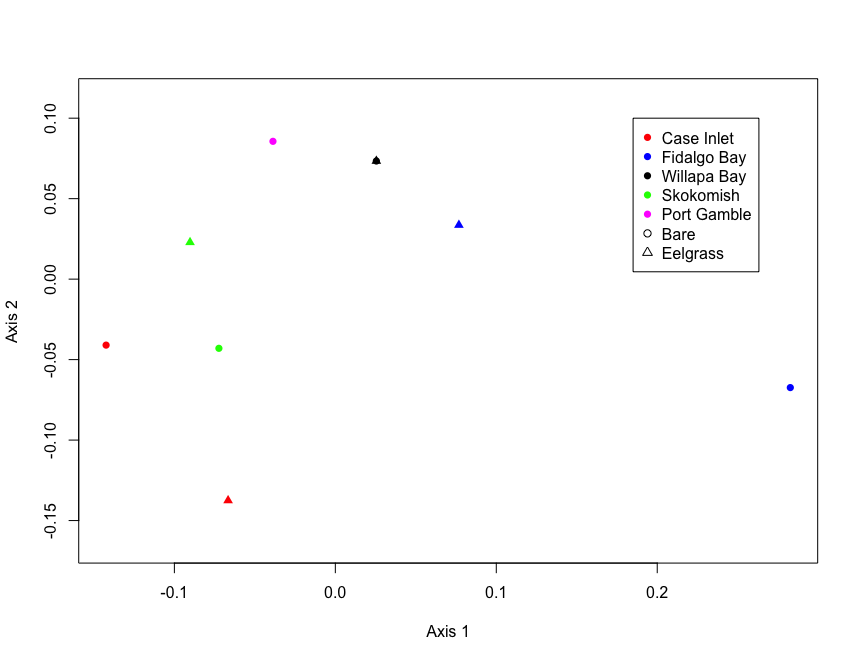
fig.nmds <- ordiplot(proc.nmds, choices=c(1,2), type='none', display='sites', xlab='Axis 1', ylab='Axis 2', cex=1)
#bare=circle
#eelgrass=triangle
#case=red
#fidalgo=blue
#willapa=black
#skokomish=green
#gamble=magenta
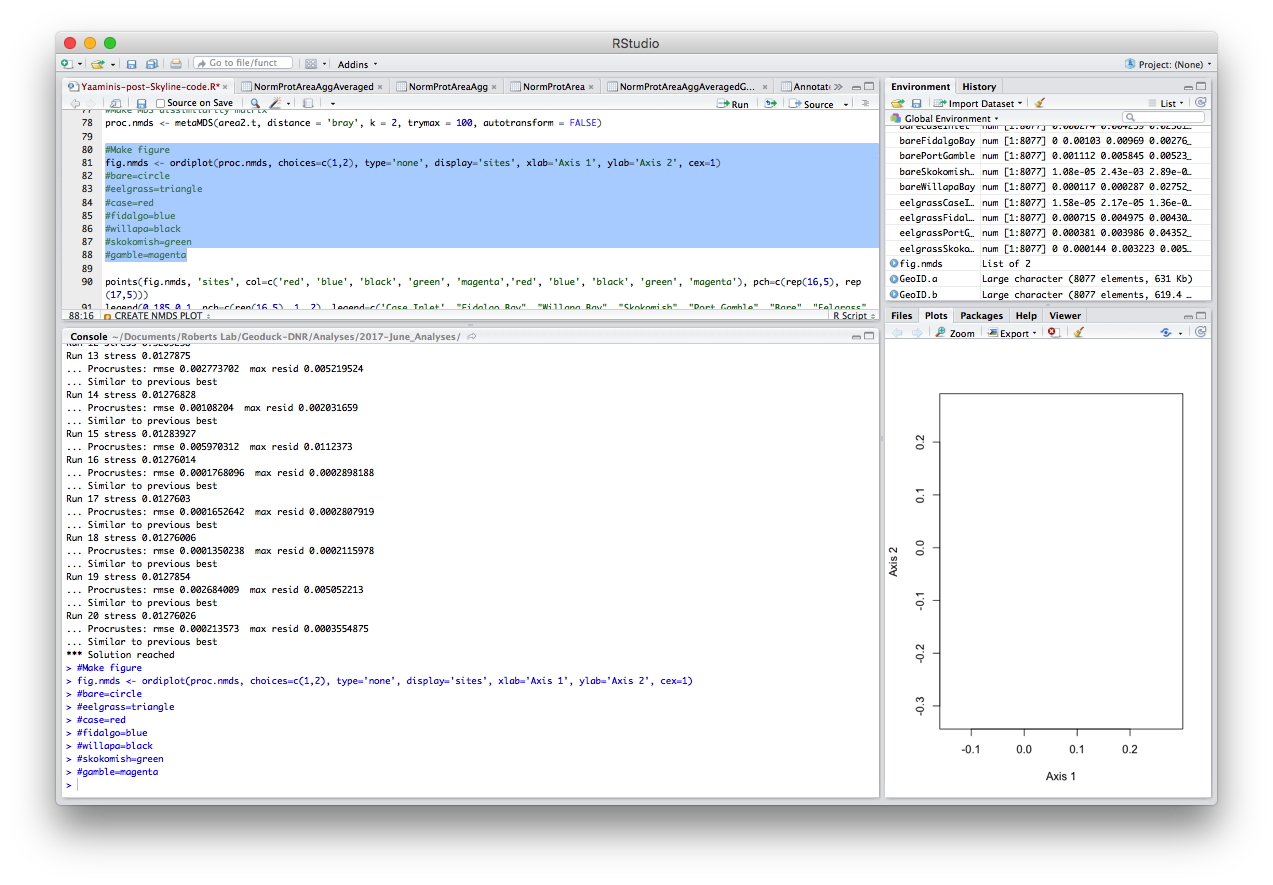
points(fig.nmds, 'sites', col=c('red', 'blue', 'black', 'green', 'magenta','red', 'blue', 'black', 'green', 'magenta'), pch=c(rep(16,5), rep(17,5)))
legend(0.185,0.1, pch=c(rep(16,5), 1, 2), legend=c('Case Inlet', "Fidalgo Bay", "Willapa Bay", "Skokomish", "Port Gamble", "Bare", "Eelgrass"), col=c('red', 'blue', 'black', 'green', 'magenta', 'black', 'black'))
FULL HEATMAP
#Install package
install.packages("pheatmap")
library(pheatmap)
#Data should be log(x) or log(x+1) transformed for this analysis. I thus invert my data (since I had normalized by TIC so values were <1), then log transform.
#Invert data then log transformation for heat map
area.protID.t.inv <- (1/area.protID.t)
is.na(area.protID.t.inv) <- sapply(area.protID.t.inv, is.infinite) # change "Inf" to "NA"
area.protID.t.inv.log <- data.trans(area.protID.t.inv, method = 'log', plot = FALSE)
ncol(area.protID.t.inv.log)
nrow(area.protID.t.inv.log)
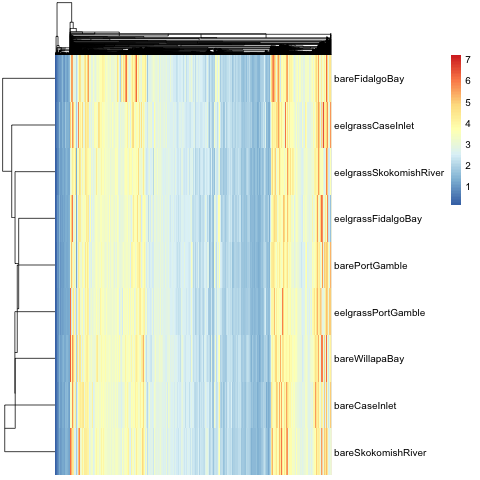
#Create heatmap
pheatmap(area.protID.t.inv.log, cluster_rows = T, cluster_cols = T, clustering_distance_rows = 'euclidean', clustering_distance_cols = 'euclidean', clustering_method = 'median', show_rownames = T, show_colnames = F)
#Export preliminary heatmap as a .png
png(filename = "2017-07-04_Heatmap-by-median.png")
pheatmap(area2.tra2, cluster_rows = T, cluster_cols = T, clustering_distance_rows = 'euclidean', clustering_distance_cols = 'euclidean', clustering_method = 'median', show_rownames = T, show_colnames = F)
dev.off()
Step 11: Calculate coefficients of variances & means, and plot. I did this to visualize how variable the proteins are- plot TBD
### Find COEFFICIENT OF VARIANCES & MEANS ###
ProtVariance <- c(var(area2.tra2[-1], area2.tra2[1], na.rm=TRUE))
ProtMeans <- c(colMeans(area2.tra2[-1], na.rm=TRUE))
ProtVar.Means <- data.frame(,y)
ProtVar.Means <- data.frame(NormProtAreaAggAveraged[1], ProtMeans, ProtVariance)
nrow(NormProtAreaAggAveraged[1])
length(ProtMeans)
length(ProtVariance)
head(ProtMeans)
ncol(ProtMeans)