
QuantSeq Library Prep test-run
Katherine suggested I work through the library prep protocol with a few samples to practice and work out kinks. From her experience, the libraries she generated later in the game were of higher quaity. I’m generating 8 test libraries - this was her recommendation based on the heavy use of 8-channel pipettes.
Following the QuantSeq manual and tips from Katherine.
Quick reference for Libary Generation

Notes from this test run:
Started on 8/20/2019
Prior to beginning, I created programs on the PTC-200 DNA Engine Cycler. Programs are labeled “Quantseq-#”, with the # corresponding to step number in the QuantSeq manual.
first strand cDNA synthesis
- Based on Katherine’s suggestions, I will generate 40 libraries at one, in 5 rows of 8 on a PCR plate. When adding solutions to each well, rather than using a single channel pipette and transferring solutions to individual wells, I’m going to use PCR strips, load enough of each solution into 8 wells (with a bit excess), and use a multichannel to distribute. This way I can hopefuly reduce time.
- Between each step I need to quickly “spin down” my PCR plate. Our benchtop centrifuge has a minimum run time of 1 minute, and with the time it takes to accelerate and decelerate total time is ~4 minutes. The “quickly spin down” instructions do not define a centrifuge speed to use, so I set it to 3 rcf based on whatt is specified in the equipmentt list - “Benchtop centrifuge (3,000 x g, rotor compatible with 96-well plates”. To reduce the amount of time to spin down plates, I should either set the speed lower, OR simply start, then stop the centrifuge manually.
RNA removal
- No notes.
second strand cDNA synthesis
- SS1 and USS are indeed viscous; hold pipette in place when pulling volumes.
- SAFE STOPPING POINT
Placed in -20C overnight.
Purificaion
(this is 1 of 2 purifications, dubbed “pre-PCR”)
- As Katherine hinted, it’s important to have a magnetic plate that fits the PCR plates used. The plates we have just have rods - no wells for plates to hold plates in place, which doesn’t work. I ordered a magnet/plate from ebay to hopefully improve the process.
- SAFE STOPPING POINT.
Placed in -20 on 8/21/19 until next step (qPCR assay).
qPCR assay for optimal # cycles
Performed on 9/3/2019
- Created a custom qPCR protocol with Sam’s help -
| qPCR Assay Mastermis Calcs | |||
|---|---|---|---|
| Item | per rxn (uL) | all rxns (inc. NTC) (uL) | all rxns * 1.1 |
| Number of samples | 1 | 9 | |
| cDNA, diluted to 19uL | 1.7 | 15.3 | 16.8 |
| PCR mix (PCR) | 7 | 63 | 69.3 |
| P7 Primer (7000) | 5 | 45 | 49.5 |
| Enzyme mix (E) | 1 | 9 | 9.9 |
| 2.5x SYBR Green I nucleic acid dye | 1.2 | 10.8 | 11.9 |
| Elution Buffer (EB) | 14.1 | 126.9 | 139.6 |
| Mastermix total vol | 28.3 | 254.7 | 280.2 |
| SYBR Green Calcs | Volumes | ||
| Stock concentration | 100 | ||
| Desired concentratino | 2.5 | ||
| Total volume needed 2.5x | 11.9 | ||
| Volume 100x | 0.2970 | ||
| Volume DMSO | 11.58 | ||
| Dilution ratio (should be 1:40) | 0.025 | ||
| Final concentration | 2.5 |
Results: Located on Owl, https://owl.fish.washington.edu/scaphapoda/qPCR_data/cfx_connect_data/ with today’s date.
No amplification :/ max RFU should be ~10x10^12, and my no-template control (NTC) has same curve as samples.

First troubleshooting step is to see if I actually synthesized cDNA - I believe I can use the Qubit for that. If yes, then I messed up the qPCR somehow (wouldn’t surprise me). Perhaps there is an issue with bubbles? Maybe BioRad settings needed be adjusted for sybr green?
I ordered a trial QuantSeq kit (n=4) for trouble shooting, and as a result spoke with the WA respresentative. Notes from our call:
- If cDNA synthesis did not occur, then I most likely had contamination with organics or salts, which can inhibit 1st strand synthesis and cause cDNA to degrade when trying to degrade RNA.
- The TurboDNase method might be an issue, since it did not include a column.
- Using a cleaner column on existing RNA may do the trick. I did order one box of Zymo Cleaner-Concentrator (n=50), so could run all my samples through this column.
- 500 ng of input RNA is not necesary (she said that “no one uses that much”). 100 ng should be adequate!
I will receive the trial kit tomorrow, and Kristy is connecting me with the tech support guy.
Update on qPCR Assay, 9/5/2019
Well, I’m an idiot, but in this case it’s a good thing. When preparing my plate for the qPCR assay I pulled my “samples” from the incorrect well, so loaded blank buffer (i.e. all NTCs) instead of samples - not surprising that I didn’t get the expected results - i.e. amplification at about 15 cycles. It is weird that there was any amplification at all, suggesting possible contamination? I re-did the qPCR assay step today. Before realizing my qPCR mistake, I also tried to quantify my ds cDNA libraries.
- Added 3uL elution buffer to all wells
- Quantified ds cDNA using the Qubit 1x dsDNA assay, and 1 uL from each of my samples.
- Results for all samples - TOO LOW. Not sure if this is expected…
- Prepared mastermix for qPCR assay just like before (calcs above), with 2 exceptsion:
- Prepared twice the amount of sybr green working solutiom - 1 uL sybr green stock + 39 uL DMSO.
- Loaded PCR plate in the AirClean PCR workstation to minimize airborn contamination.
- Spun PCR plate down after preparing to remove bubbles. This was harder than anticipated - the ~3,000 rcf speed did not remove bubbles, so I went as high as the benchtop centrifuge would allow - 4,000 for 3 minutes. According to the centrifuge manual it should go as high as ~10,000 rcf, so not sure why I kept getting an error message at speeds above 4k.
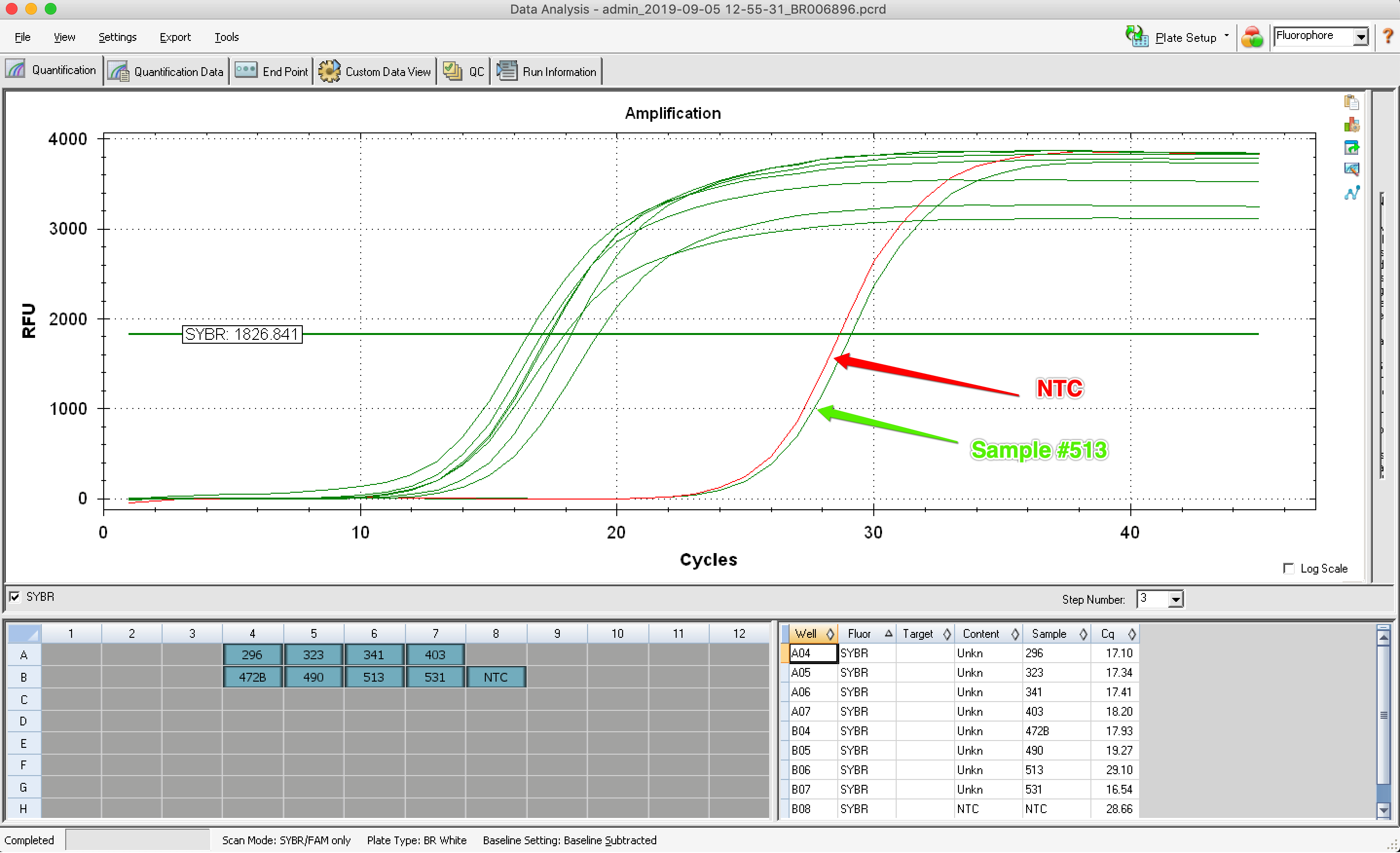
Results: 7 out of 8 samples started to amplifly at the expected cycle! One did not, producing an amplification curve similar to the NTC.
| Well | Fluor | Content | Sample | End RFU |
|---|---|---|---|---|
| A04 | SYBR | Unkn | 296 | 3531 |
| A05 | SYBR | Unkn | 323 | 3829 |
| A06 | SYBR | Unkn | 341 | 3848 |
| A07 | SYBR | Unkn | 403 | 3850 |
| B04 | SYBR | Unkn | 472B | 3118 |
| B05 | SYBR | Unkn | 490 | 3253 |
| B06 | SYBR | Unkn | 513 | 3735 |
| B07 | SYBR | Unkn | 531 | 3784 |
| B08 | SYBR | NTC | NTC | 3841 |
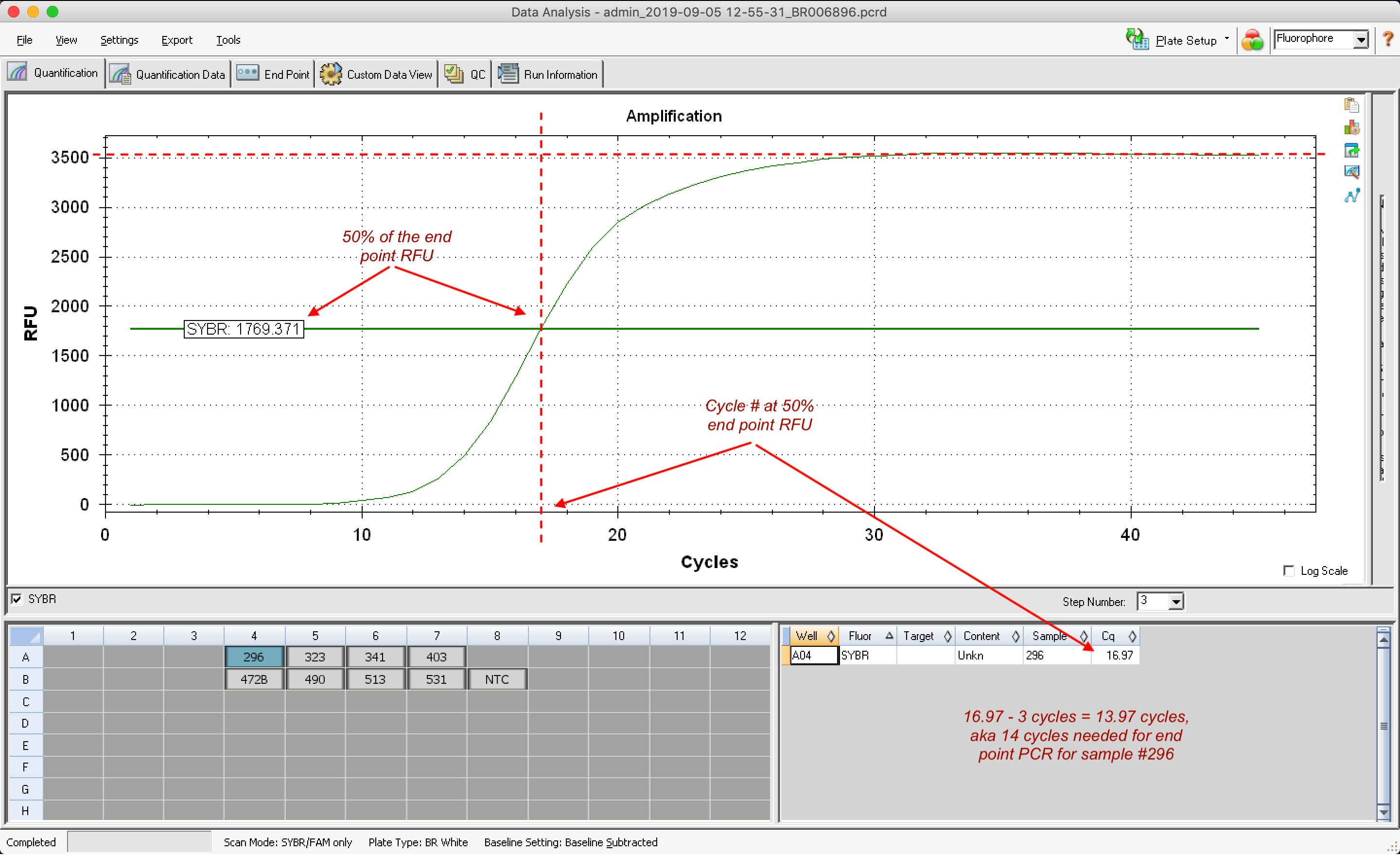
To calculate the optimal number of cycles for endpoint PCR when amplifying/adding adapters to my ds cDNA I do the following:
- Determine endpoint RFU, aka RFU value at plateau
- Calculate 50% of endpoint RFU
- Identify cycle # at 50% endpoing RFU
- Subtract 3 cycles
Katherine advises that I should round down if the calculated cycle number is a decimal, to avoid overcycling.
| Cohort | Treatment | Tissue | RNA Sample no. | RFU @ endpoint | 50% max | No. Cycles @ 50% | No. Cycles @ 50% minus 3 cycles | Cycles, round down |
|---|---|---|---|---|---|---|---|---|
| Dabob Bay | 6 Low High | Ctenidia | 296 | 3530 | 1,765 | 16.97 | 13.97 | 14 |
| Oyster Bay | 6 Ambient | Ctenidia | 323 | 3829 | 1,915 | 17.52 | 14.52 | 14 |
| Fidalgo Bay | 6 Ambient | Ctenidia | 341 | 3848 | 1,924 | 17.59 | 14.59 | 14 |
| Dabob Bay | 10 Ambient | Larvae | 403 | 3850 | 1,925 | 18.38 | 15.38 | 15 |
| Fidalgo Bay | 6 Low | Larvae | 472b | 3118 | 1,559 | 17.27 | 14.27 | 14 |
| Oyster Bay C1 | 10 Ambient | Larvae | 490 | 3253 | 1,627 | 18.81 | 15.81 | 15 |
| Oyster Bay C1 | 6 Ambient | Larvae | 513 | 3735 | 1,868 | 29.17 | 26.17 | NA |
| Oyster Bay C2 | 10 Ambient | Larvae | 531 | 3784 | 1,892 | 16.68 | 13.68 | 13 |
| NTC | 3840 | 1,920 | 28.20 | 25.20 | NA |